Dogs teach us so many things. My first time dog sitting, I learned the difference between an asthmatic pug wheezing and preparing to vomit. Two days in and I could go from a dead sleep to holding him over the tile in three seconds. Sometimes, dogs vomit because they decided to eat the outcome of the last “why did the chicken cross the road joke”, and then they’re fine. Sometimes, dogs vomit because Midwestern Pet food sold you dog food with aflatoxin in it, and then they die. In that case, dogs teach us not to trust food with “holistic” in the name.

Figure 1. Vomiting is just (aflo)toxins leaving the body
The FDA got involved after more that 110 pets died of aflatoxin poisoning after eating certain pet food manufactured by Midwestern Pet Foods. Aflatoxins are mycotoxins, toxic compounds produced by mold1 . Aflatoxin poisoning causes the overactivation of the inflammatory response, damaging the liver. This causes side effects ranging from sluggishness and vomiting to convulsions and death. Aflatoxins can also be inhaled and absorbed through the skin, mainly a concern for the workers handling infected food grains2. Contaminated dog food isn’t likely to hurt a human scooping it out with their hands, but I’d recommend not snorting the powder at the bottom of the bag.
How does a well established3, multimillion dollar company end up with aflatoxin, a very well known and extremely dangerous toxin, in its products? Midwestern pet food has a quality control department, and they tested for aflatoxins, presumably only releasing lots within allowable limits. Switching suppliers and disposing of tainted raw materials that failed aflatoxin testing is way cheaper than decontaminating every piece of equipment that it comes into contact with in the facility. Managers with site-wide production bonuses are VERY protective of their manufacturing lines, because a weeklong shutdown to disassemble and disinfect every piece of equipment that tainted materials came into contact with can wreck a plants profits for the year. So they wouldn’t allow contaminated materials to be used on their equipment.
The company as a whole has no incentive to sell tainted batches, because even trashing a million dollar batch is cheaper than the negative press and government scrutiny that comes with a giant public health hazard. And yet, the FDA found almost 30 times the allowable limit of aflatoxin in their products, leading to a very public warning letter stating that there is a “reasonable possibility that a regular diet of such food will be fatal or injurious to the health of the pet.” Hard to PR your way out of that.
Aflatoxin was found in products across FOUR separate plants. This was not a single contamination event, but an inadequate company wide policy. A submitted food safety plan (the same for all affected plants) identified aflatoxin in incoming corn as a possible safety hazard. Destroying Aflatoxins requires temperatures of 300°C, so instead of mechanically destroying any possible aflatoxin, the decision was made to test all materials for aflatoxin contamination. The warning letter states Midwestern pet food “failed to follow proper sample preparation procedure … that your facilities reported as being followed. This led to potentially inaccurate analyses and test results for sampled products.
Aflatoxins are small (molecular weight < 1000), and have diverse chemical structures, with four major types. They also are tested for in a variety of different matrixes (the sample being tested, such as corn, wheat or dog food). This makes them hard to create robust testing methods for.
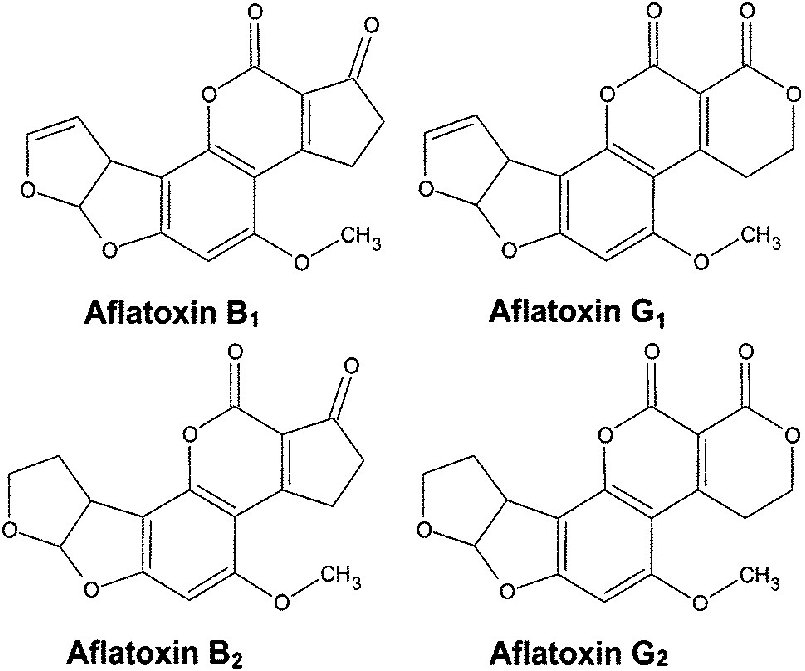
Figure 2. Every quality control scientist’s worst nightmare
Because aflatoxins are present in complex matrixes, sample preparation is one of the most important steps in their detection. A sample of dog food would be ground, mixed with the extraction solution (70:30 Methanol:Water), and then filtered and tested. Normally, you would compare the sample that you are testing to a sample that you know has aflatoxins in it, that underwent the same treatment as the sample. However, you can’t buy dog food with aflatoxin in it to compare your sample to. Instead, you buy pure aflatoxin and test that along with your sample. But if the sample preparation process doesn’t successfully extract aflatoxin from dog food, then there is no aflatoxin in the sample tested, but there could be aflatoxin in the dog food. That test would be negative for every product tested, even if there was aflatoxin in the dog food.
To prevent this, test methods should be validated (proven to work) on each individual product, proving that the sample preparation method actually removes any aflatoxins from the sample. But this is time consuming and expensive. To prove the sample prep could remove aflatoxin from dog food, a sample of each product would have to be incubated with a specific species of mold (so that it would have aflatoxins in it), and then tested repeatedly. Buying a kit off of the internet is fast and cheap. Proving it actually detects aflatoxins in your products is time consuming and expensive.
Of course, then the FDA tells the entire world “your facilities failed to follow proper sample preparation procedures… that your facilities reported as being followed. This led to potentially inaccurate analyses and test results for sampled products.” It doesn’t take a consultant to tell you that validating your test methods is looking like a better and better deal.( but if you want to pay me six figures to tell you shit you already know, I could make some time)

Figure 3. What I do all day
It’s tempting to blame the dead dogs on lazy quality control testing leading to false negatives. But contaminated products are never simply a failure of quality control testing, but a failure of each step in the supply chain. Manufacturing didn’t mechanically destroy any possible aflatoxins in the raw materials, so they ended up in the products. By the time quality control finds (or doesn’t find) toxins in your finished product, you have bigger problems than just discarding one batch and papering over the holes in your process. Companies have to go back to their manufacturing process and figure out how to prevent issues from occurring in the first place.
I’m not trying to rag on Midwestern pet food specifically. Companies (even ones that don’t lowkey murder puppies) sometimes have contamination issues. God knows I’ve given myself food poisoning more times than I’ll admit to. But I’m poor, and Midwestern is worth 500 million dollars. I wouldn’t eat the same sketchy takeout more than once, but Midwestern kept producing batches after they knew they had a problem, without updating their manufacturing processes. Midwestern pet food eventually pulled multiple products over several months, each with aflatoxin contamination. Nobody wants to hear that the site is going to have to shut down for a week to clean every piece of equipment that came into contact with a contaminated batch, but nobody wants a dead dog either.
TL;DR: validate your methods or the puppy gets it

Figure 3. Look at him!
1Specifically, Aspergillus flavus, which grows on grains such as corn, wheat and peanuts which are used in dog food
2If Midwestern thought recalls and pet death lawsuits were expensive, wait until OSHA gets ahold of this
3Midwestern was founded in 1926